"T'i njohim sëmundjet e rralla": Sindromi Ohtahara
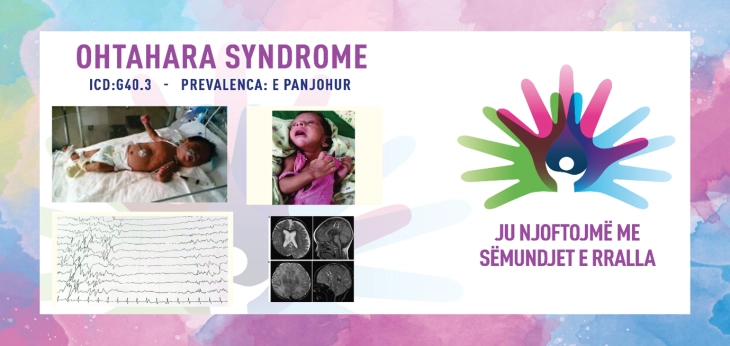
SINDROMI OHTAHARA/OHTAHARA SYNDROME/EARLY INFANTILE DEVELOPMENT & EPILEPTIC ENCEPHALOPATHY (EIDEE)
ICD-10 G40.3
PREVALENCA - e panjohur.
INCIDENCA - sindroma Ohtahara shihet në afërsisht 1/50 000 lindje në Britaninë e Madhe deri në 1/100 000 lindje në Japoni.
HYRJE
Sindroma Ohtahara është formë e rrallë e epilepsisë e karakterizuar nga kriza dhe vonesa në zhvillim që zakonisht ndodhin në tremujorin e parë të jetës, por disa shfaqen në javët e para pas lindjes (kryesisht në 10 ditët e para). Disa raste shkaktohen nga një anomali e trurit, çrregullimi metabolik ose mutacioni i gjenit, ndërsa për raste të tjera nuk ka ndonjë shkak të njohur. Vajzat dhe djemtë janë të prekur në mënyrë të barabartë. Foshnjat më së shpeshti kanë sulme tonike (ngurtësi muskulore, shikim te fiksuar lart, bebëza të zgjeruara dhe frymëmarrje të ndryshuar), por ata gjithashtu mund të përjetojnë kriza fokale (që prekin vetëm një zonë ose anë të trurit) dhe rrallë, konvulsione mioklonike (konvulsione të papritura ose spazma të ekstremitetit sipërm te trupit, krahët dhe këmbët). Fëmijët me këtë çrregullim mund të kenë më shumë se një lloj konvulsionesh dhe konvulsionesh simetrike që mund të ndodhin në grupe ose individualisht dhe mund të zgjasin deri në 10 sekonda.
ETIOLOGJIA
EIDEE mund të jetë rezultat i etiologjive të ndryshme, madje 80% e rasteve lidhen me anomalitë strukturore të trurit. Disa raste janë për shkak të çrregullimeve metabolike (mungesa e citokrom C-oksidazës, mungesa e karnitine palmitoyl transferazës II) ose keqformime të trurit (porencefalia, hemigalencefalia) të cilat gjithashtu mund të jenë me origjinë gjenetike. Variantet gjenetike të EIDEE shoqërohen me mutacione në gjene të caktuara, si ARX (Xp22.13), CDKL5 (Xp22), SL25A22 (11p15.5) dhe STXBP1 (9q34.1), ndër të tjera. Anomalitë gjenetike mendohet se çojnë në EIDEE sepse ato shoqërohen me mosfunksionim neuronal ose disgenezë të trurit. Shkaqet metabolike mund të përfshijnë hiperglicinemi joketonike, çrregullime të aminoacideve organike, çrregullime të ciklit të uresë, çrregullime mitokondriale, çrregullime të piridoksinës dhe piridoksal-5-fosfatit, mungesës së oksidazës së sulfiteve dhe sindromës Menkes (sindroma Zellweger). Në disa, EIDEE mund të përparojë në sindromën West (ndërmjet moshës 2 deri në 6 muaj) dhe më vonë në sindromën Lennox-Gastaut. Disa variante gjenetike manifestohen me shenja shtesë, të tilla si lëvizjet diskinetike dhe një fenotip atipik të sindromës Rett.
SIMPTOMAT DHE DIAGNOSTIKIMI
Të porsalindurit me sindromën Ohtahara zhvillojnë konvulsione të shpeshta në 3 muajt e parë të jetës. Zakonisht shfaqen në 2 javët e para pas lindjes. Këto sulme ndonjëherë janë të vështira për t'u kontrolluar. Ata shpesh kanë nevojë për ilaçe kundër konvulsioneve. Të porsalindurit kanë reflekse të dobëta thithëse, hipotoni dhe paraqiten me kriza të gjeneralizuara. Modeli i këtyre spazmave mbetet i pandryshuar gjatë zgjimit dhe gjumit, ndërsa ato mund të ndodhin qindra herë në ditë. Zakonisht zgjasin vetëm disa sekonda dhe mund të marrin formën e mpirjes në krahë dhe/ose këmbë, prekin njërën ose të dyja anët e trupit, ndodhin vetëm ose në grupe, ndodhin kur foshnja është zgjuar ose në gjumë.
Lloje të tjera konvulsionesh, duke përfshirë konvulsione të përgjithësuara toniko-klonike (kriza që prekin të dyja anët e trurit dhe fillojnë me një humbje të papritur të vigjilencës, e ndjekur nga ngurtësim i muskujve dhe lëvizje të vrullshme), kriza motorike fokale (mund të shkaktojnë lëvizje jonormale vetëm në një pjesë të trupit) dhe hemikonvulsione, vërehen në 1/3 e rasteve. Foshnjat me këtë sindromë kanë vonesa në zhvillim. Ky mund të jetë rezultat i shkakut themelor të gjendjes së tyre të kombinuar me konvulsione të vazhdueshme. Ata shpesh nuk arrijnë zhvillimin në të njëjtën moshë si foshnjat e tjera. Ata gjithashtu mund të kenë rezultate jonormale në një ekzaminim neurologjik. Simptomat e zhvillimit mund të jenë të pranishme përpara se të fillojnë krizat dhe mund të përkeqësohen me rritjen e frekuencës së krizave.
Diagnoza bazohet në të dhënat klinike dhe elektroencefalografike. Elektrencefalogrami karakteristik (EEG) tregon një model të ndezur të shtypur që ndodh me fillimin e spazmave dhe përbëhet nga breshëri të thumbave me amplitudë të lartë dhe thumba të alternuara me periudha të një vije bazë të tensionit të ulët. Ky model EEG është i vazhdueshëm dhe mbetet i pandryshuar si gjatë zgjimit ashtu edhe gjatë gjumit. Anomalitë strukturore shpesh mund të shihen në MRI të kafkës. Në disa të porsalindur kryhen analiza laboratorike për të identifikuar shkakun e sindromës, si dhe analiza të urinës që synojnë çrregullimet metabolike. Testet gjenetike përfshijnë mikrogrumbullimet kromozomale, panelet gjenetike të epilepsisë dhe sekuencën e tërë ekzomit që mund të ndihmojnë në gjetjen e shkakut të sindromës Ohtahara.
PASOJAT
Prognoza është mjaft e keqe dhe vdekja zakonisht ndodh në fëmijëri (50% para moshës 2 vjeçare). Të mbijetuarit kanë dëmtime të rënda psikomotore me konvulsione të vazhdueshme. Ndërsa rriten, disa fëmijë zhvillojnë çrregullime të tjera të epilepsisë si sindroma West dhe sindroma Lennox-Gesto. Anëtarët e familjes së foshnjave mund të konsultohen me një ekip të kujdesit paliativ pasi simptomat mund të përkeqësohen ose zhvillohen. Vdekja është shpesh për shkak të barrës së konfiskimeve, pneumonisë ose komplikimeve të tjera të dëmtimit motorik.
TRAJTIMI
Duke qenë se nuk ka kurë për EIDEE, pacientët kanë nevojë për monitorim dhe kujdes të vazhdueshëm. Trajtimi mund të përfshijë kortikosteroide, një dietë ketogjenike (me yndyrë të lartë, me pak karbohidrate) dhe terapi fizike, të të folurit dhe profesionale. Kirurgjia e epilepsisë, në rastet kur është i pranishëm një lezion fokal i trurit (dëmtim ose zhvillim jonormal i një zone/anës së trurit), mund të jetë i dobishëm sepse barnat antiepileptike, si benzodiazepinat, valproati, levetiracetami, zonisamidi dhe fenobarbitali, kanë treguar sukses të kufizuar në kontrollin e krizave si dhe piridoksinës. Dieta ketogjenike është treguar se tregon njëfarë suksesi në kontrollin e konvulzioneve. Pacientët me EIDEE me anomali të caktuara strukturore përfitojnë nga ndërhyrja kirurgjikale, nëse ajo është e njëanshme. Shumica e rasteve të EIDEE janë sporadike (mutacione "de novo"), kështu që këshillimi gjenetik rekomandohet për të informuar prindërit se rreziku i tyre për të pasur fëmijë të mëtejshëm me EIDEE është i ulët.
Foto: Ilustrim











